ソニケーションクロマチン免疫沈降プロトコール
次の製品の専用プロトコールです: SimpleChIP® Plus Sonication Chromatin IP Kit #56383.
I. 組織のクロスリンクおよびサンプル調製
組織を採取する際、脂肪や壊死組織などの不要な部分を取り除きます。組織を処理し直ちにクロスリンクするか、使用するまでドライアイスで凍結して保存してください。最適なChIP結果を得るため、各免疫沈降あたり25 mgの組織を使用します。クロマチンの断片化および濃度の解析 (セクションIV) とインプットクロマチンとして使用するため、追加で5 mgの組織を用意してください。クロマチン収量は、組織の種類により異なりますが、一部の組織では各免疫沈降あたり25 mgを超える量が必要となる可能性があります。
クロマチン調製1回に必要な量として、組織100 - 150 mgを想定しています。推奨の組織量を用いることで、いくつかの組織タイプでクロマチン収量が低くなる問題を回避するとともに、ソニケーション処理による効率的なクロマチン断片化を行うことが可能となります。組織ごとに予測されるクロマチン収量については、Appendix Aを参照してください。
実験開始前の準備:
- 200X Protease Inhibitor Cocktail (PIC) #7012 とGlycine Solution (10X) #7005 を取り出し、室温に戻してください。PICが完全に溶けていることを必ず確認してください。
- クロマチン調製1回あたり、リン酸緩衝生理食塩水 (PBS) 3 mL + 200X PIC 15 µLの混合液を調製し、氷上においてください。
- クロマチン調製1回あたり、1X ChIP Sonication Cell Lysis Buffer 1 mL (2X ChIP Sonication Cell Lysis Buffer #96529 0.5 mL + 水 0.5 mL) + 200X PIC 5 µLの混合液を調製し、氷上に置きます。
- クロマチン調製1回あたり、37%ホルムアルデヒド28 µLを用意し、室温に置いてください。代わりに、メタノールを含まない16%ホルムアルデヒド62.5 µLを使用することもできます。使用期限内の新しいホルムアルデヒドを使用してください。
- 新鮮組織または凍結組織サンプルを計量してください。クロマチン調製1回あたり、100-150 mgの組織を使用してください。
- 組織サンプルをペトリ皿に入れ、清潔な解剖用メスまたは剃刀の刃を用いて、1-2 mm角になるまで細かく切り刻んでください。シャーレは氷上に置いてください。タンパク質分解を防ぐため、組織をよく冷やす必要があります。
- ミンスした組織を15 mLのコニカルチューブに入れ、クロマチン調製ごとに氷冷したPBS + PIC混合液1 mLを加えてください。
- タンパク質をDNAにクロスリンクするため、PBS + PIC混合液1 mLに対し、37%ホルムアルデヒド28 µLまたは16%メタノールフリーホルムアルデヒド62.5 µLを加え、室温で少なくとも10分間置いてください。最終的なホルムアルデヒド濃度は1%です。ヒストン修飾に対するChIPでは10分間の固定で十分ですが、転写因子に対するChIPでは10 - 30分間固定することを推奨します。また、転写コファクターのChIPでは、30分間固定することを推奨します (Appendix Bの図7を参照してください)。
- PBS + PIC混合液1 mLに対し、10X Glycine #7005 100 µLを加えてクロスリンクを停止させてください。混和し、氷上で5分間インキュベートしてください。
- 組織を、4℃、1,200 x gで5分間遠心分離してください。
- 上清を取り除き、クロマチン調製ごとに氷冷したPBS + PIC混合液1 mLで洗ってください。
- 4℃、1,200 x gで5分間遠心分離してください。
- ステップ7と8をもう一度繰り返してください。
- 上清を取り除き、クロマチン調製ごとに1X ChIP Sonication Cell Lysis Buffer + PIC混合液1 mLを入れ、組織を再懸濁してください。
- 先端を切ったピペットチップを用いて、組織懸濁液を塊ごとDounce Homogenizerに入れてください。
- Tightのペストル (タイプA) を用いて、20回または塊が見えなくなるまでストロークし、組織を破砕してください。
- 細胞懸濁液を1.5 mLのチューブに移し、直ちに細胞核の調製とクロマチン断片化に進んでください (セクションIII)。
II. 培養細胞のクロスリンクおよびサンプル調製
最適なChIPの結果を得るため、免疫沈降1回あたり約4 x 106 細胞を使用します。HCT 116細胞の場合、増殖培地20 mLで90%コンフルエントに培養した15 cm培養ディッシュ1枚分の1/3量に相当します。クロマチン断片化および濃度の解析、およびインプットクロマチンとして使用するため、追加で1 x 106 個の細胞を調製してください (セクションIV)。
クロマチン調製1回に必要な量として、1 x 107-2 x 107個の細胞を想定しています。推奨の細胞数を用いることで、いくつかの細胞種でクロマチン収量が低くなる問題を回避するとともに、ソニケーション処理による効率的なクロマチン断片化を行うことが可能となります。
実験開始前の準備:
- 200X Protease Inhibitor Cocktail (PIC) #7012 およびGlycine Solution (10X) #7005 を取り出し室温に戻してください。PICが完全に溶けていることを必ず確認してください。
- 15 cmディッシュ1枚あたり、Phosphate Buffered Saline (PBS) 2 mL + 200X PIC 10 μLを調製して、氷上に保持してください。
- 15 cmディッシュ1枚あたりPBS 40 mLを調製し、氷上に保持してください。
- 15 cmディッシュ1枚分の細胞に対して、37%ホルムアルデヒド540 μLを調製し、室温に保持してください。あるいは、1.25 mLの 16%メタノールフリーのホルムアルデヒドを使用することもできます。使用期限内の新しいホルムアルデヒドを使用してください。
- クロマチン調製1回あたり、1X ChIP Sonication Cell Lysis Buffer 1 mL (2X ChIP Sonication Cell Lysis Buffer #96529 0.5 mL + 水0.5 mL) + 200X PIC 5 µLの混合液を調製してください。
- タンパク質をDNAにクロスリンクするため、20 mLの培地を含む15 cmの培養ディッシュに、37%ホルムアルデヒド540 μLまたは16%メタノールフリーホルムアルデヒド1.25 mLを加えてください。手早くディッシュを傾けて混ぜ合わせ、室温で10分間インキュベートしてください。最終的なホルムアルデヒド濃度は1%です。ホルムアルデヒドを加えると、培地の色が変化することがあります。
- 10X Glycine Solution 2 mLを、培地が20 mL入ったそれぞれの15 cmディッシュに加え、手早く回して混ぜ合わせ、室温で5分間インキュベートしてください。glycineを加えると、培地の色が変化することがあります。
- 浮遊細胞の場合:
- 50 mLのコニカルチューブに移し、4℃、1,000 x gで5分間遠心分離し、20 mLの氷冷したPBSでペレットを2回洗ってください。PBSを取り除き、ステップ3bへ進んでください。あるいは、細胞ペレットをドライアイス等で凍結し、使用するまで-80℃で保存してください。
- クロマチン調製1回あたり、細胞数が最大2 x 107個/mLとなるように、1X ChIP Sonication Cell Lysis Buffer + PIC混合液で再懸濁し、直ちに細胞核の調製およびクロマチンの断片化に進んでください (セクションIII)。
- 接着細胞の場合:
- 培地を除去し、氷冷した1X PBS 20 mLで2回洗い、毎回洗いの液を完全に取り除いてください。
- 氷冷したPBS + PIC混合液2 mLを、それぞれの15 cmのディッシュに加えてください。細胞を掻き取り、バッファーに懸濁してください。すべてのディッシュから集めた細胞をコニカルチューブ (15 mL) 1本に集めてください。
- 細胞を4℃にて1,000 x gで5分間遠心分離してください。PBSを取り除き、ステップ4dへ進んでください。あるいは、細胞ペレットをドライアイスで凍結し、使用するまで-80℃で保存することが可能です。
- クロマチン調製1回あたり、細胞数が最大2 x 107個/mLとなるように、1X ChIP Sonication Cell Lysis Buffer + PIC混合液で再懸濁し、直ちに細胞核の調製およびクロマチンの断片化に進んでください (セクションIII)。
III. 細胞核の調製とクロマチン断片化
クロマチン調製1回に必要な量として、100-150 mgの組織または1 x 107-2 x 107個の培養細胞を想定しています。複数回分のクロマチン調製を同時に行うことが可能ですが、バッファー量をそれに合わせてスケールアップし、ソニケーション処理は1 mLのサンプルスケールで行ってください。ソニケーション時の細胞数と液量は、得られるクロマチン断片のサイズに影響します。
実験開始前の準備:
- 200X Protease Inhibitor Cocktail (PIC) #7012を取り出し、室温に戻してください。使用前に完全に解凍されていることを確認してください。
- クロマチン調製1回あたり、1X ChIP Sonication Cell Lysis Buffer 1 mL (2X ChIP Sonication Cell Lysis Buffer #96529 0.5 mL + 水0.5 mL) + 200X PIC 5 µLの混合液を調製してください。
- クロマチン調製1回あたり、ChIP Sonication Nuclear Lysis Buffer #28778 1 mL + 200X PIC 5 µLの混合液を調製してください。
- セクションIまたはセクションIIで調製した1X ChIP Sonication Cell Lysis Buffer + PIC混合液で懸濁した細胞サンプルを、氷上で10分間インキュベートしてください。
- 5,000 x gで5分間4℃で遠心分離してください。上清を取り除き、クロマチン調製ごとに氷冷した1X ChIP Sonication Cell Lysis Buffer + PIC混合液 1 mLを加え、ペレットを再懸濁してください。
- 細胞懸濁液を氷上で5分間インキュベートしてください。5,000 x gで5分間4℃で遠心分離してください。クロスリンクした細胞は、ソニケーション処理するまでは完全に溶解しないことにご留意ください。
- クロマチン調製ごとに、細胞を氷冷したChIP Sonication Nuclear Lysis Buffer #28778 + PIC混合液1 mLで懸濁し、氷上で10分間インキュベートしてください。ソニケーションに適切なサイズのチューブに、細胞懸濁液1 mLを移してください。クロスリンクした細胞および核は、ソニケーション処理するまでは完全に溶解しないことにご留意ください。
- ソニケーションによるクロマチンの断片化:超音波処理の条件は、ソニケーターごとに出力設定と超音波の処理時間をテストすることによって、経験的に決定する必要があります。最適な超音波処理条件では、クロマチン断片の60-90%が1 kb未満のサイズとなります。クロスリンクの時間が長いと、1 kb未満の断片の割合が30-60%に減少する可能性があります (Appendix Bの図7、Appendix Cの図8を参照)。過剰な超音波処理は、クロマチンの苛酷な処理によってシグナルが減少または消失する原因となり得るため、推奨されるサイズのクロマチン断片が得られる必要最低限の回数の超音波処理を行うようにしてください。
- 各ソニケーションサンプルに対し、100-150 mgの組織または1 x 107-2 x 107個の細胞を、1 mLのChIP Sonication Nuclear Lysis Bufferに懸濁することを推奨しています。組織量が多かったり、細胞密度が高かったりすると、クロマチン断片化の効率が低下する恐れがあります。
- Branson Digital Sonifier D250 probe sonicatorを1/8-inch Micro Tipとともに使用し、50%の出力設定で1秒間ON + 1秒間OFFのサイクルを計8分間 (ソニケーション処理は4分間) 行うと、最適なクロマチンの断片化が起こり、クロマチン免疫沈降の効率も良くなります。
- ソニケーション処理中は、クロマチンサンプルの入ったチューブを氷水中に置き、常に冷やすようにしてください。ソニケーターのプローブがチューブの底や側面に触れないようにしてください。ソニケーション中にクロマチンサンプルが泡立った場合は、一旦停止し、チューブの位置を調整してください。
- 21,000 x gで10分間、4°Cで遠心分離して、ライセートを清澄化してください。
- 上清を新しいチューブに移してください。これが、クロスリンクしたクロマチン調製液です。直ちに免疫沈降に使用するか、使用するまで‐80℃で保存してください。50 μLのクロマチン調製液を、クロマチンの断片化および濃度の解析 (セクションIV) に使用します。
IV. クロマチンの断片化および濃度の解析 (推奨ステップ)
- クロマチンサンプル 50 μL (セクションIIIのステップ7) に、Nuclease-free water 100 µL、5 M NaCl #7010 6 µL、RNAse A #7013 2 µLを加えてください。ボルテックスで混和し、37℃で30分間インキュベートしてください。
- RNase Aで処理した各サンプルに、Proteinase K #10012 2 μLを加えてください。ボルテックスにより混和し、65℃で2時間インキュベートしてください。
- セクションVIIに記載されているとおり、DNA精製用スピンカラムを用いてDNAをサンプルから精製してください。
- DNA精製後、各サンプル10 μLを1%アガロースゲルで電気泳動し、DNA断片のサイズを測定してください。DNAはスメア状に、200 bp-数 kbの範囲で観察されることが予想されます (Appendix Cの図7を参照)。全DNA断片の約60-90%が、1 kb未満となっているはずです。ソニケーション条件の最適化については、Appendix Bを参照してください。
- 分光光度計を用いてDNA濃度を測定してください。DNA濃度が50 - 200 µg/mLになることが理想です。
V. クロマチン免疫沈降 (ChIP)
最適なChIPの結果を得るために、1回の免疫沈降あたり約5-10 μgのソニケーション処理されたクロスリンククロマチンを使用してください (セクションIVで濃度決定)。これは、破砕した組織25 mgまたは4 x 106個の培養細胞から調製したクロマチンサンプル100 μLに相当します。一般的には、抗体を加える前にクロマチンサンプル100 μLを1X ChIP Buffer 400 μLで希釈しますが、濃度が低く、100 µL以上のクロマチン調製液が必要になる場合は、1X ChIP Bufferを用いてクロマチンサンプルを1:4に希釈してください。Protein G magnetic beadsを追加で加える必要はありませんが、ビーズとのインキュベーション時間の延長は有効です。
実験開始前の準備:
- 200X Protease Inhibitor Cocktail (PIC) #7012を取り出し、室温に戻してください。PICが完全に溶けていることを必ず確認してください。
- 10X ChIP Buffer #7008を取り出して室温に戻し、SDSが完全に溶解していることを確認してください。
- 断片化したクロマチン調製液 (セクションIIIのステップ7) を溶かし、氷上に置いてください。
- low salt washの調製:免疫沈降1回あたり、1X ChIP Buffer 3 mL (10X ChIP Buffer #7008 300 μL + 水2.7 mL) を調製し、氷上に保持してください。
- high salt washの調製:免疫沈降1回あたり、1X ChIP Buffer 1 mL (10X ChIP Buffer #7008 100 μL + 水900 μL) + 5M NaCl #7010 70 μLの混合液を調製し、氷上に保持してください。
- 1本のチューブに、免疫沈降の予定回数分に十分な1X ChIP Buffer + PIC混合液を用意してください。免疫沈降1回あたり計500 μLとなるように、クロマチンサンプル (DNA 5-10 µg) を1X ChIP Buffer + PIC混合液で希釈してください。効率的な免疫沈降のために、クロマチンを1X ChIP Bufferで1:4以上の希釈率で希釈してください。調製したチューブを氷上に保持してください。ポジティブコントロールのHistone H3 (D2B12) XP® Rabbit mAb #4620およびネガティブコントロールのNormal Rabbit IgG Antibody #2729 の分も忘れずに用意してください。
- 希釈したクロマチン10 μLを遠心分離用チューブに移してください。これが2%インプットサンプルです。使用するまで-20℃で保存してください (セクションVIのステップ1で使用します)。
- 免疫沈降1回あたり、500 μLの希釈したクロマチンを1.5 mLの遠心分離用チューブに移し、免疫沈降用抗体を加えてください。必要な抗体量は免疫沈降ごとに異なるため、メーカーの推奨に従って使用してください。ポジティブコントロールのHistone H3 (D2B12) XP®Rabbit mAb #4620については、10 μLを免疫沈降に使用してください。ネガティブコントロールのNormal Rabbit IgG #2729については、1-2 μL (1-2 μg) を免疫沈降に使用してください。ローテーターを用いて、各免疫沈降サンプルを4℃で4時間から一晩インキュベートしてください。
注意:Cell Signaling Technologyのほとんどの抗体は、1-2 μgで使用することで最適な結果が得られます。濃度の異なる複数のサンプルがある場合は、最も高い抗体濃度にネガティブコントロールNormal Rabbit IgG #2729の濃度を合わせて使用してください。
- ChIP Grade Protein G Magnetic Beads #9006を穏やかにボルテックスし、再懸濁してください。直ちに30 μLのProtein G Magnetic Beadsを免疫沈降サンプルに加え、ローテーターを用いて4℃で2時間インキュベートしてください。あるいは、Protein G Magnetic Beadsをステップ3で加えることもできますが、バックグラウンドが高くなる可能性があります。
- 各免疫沈降ごとにチューブをMagnetic Separation Rackにセットし、Protein G Magnetic Beadsを分離させてください。溶液が澄むまで1-2分間待ち、上清を注意深く取り除いてください。
- low salt wash 1 mLを加えてProtein G Magnetic Beadsを洗い、4℃で5分間ローテーターでインキュベートしてください。ステップ6と7を更に2回繰り返し、計3回low salt washで洗ってください。
- high salt wash 1 mLをBeadsに加え、4℃で5分間ローテーターを用いてインキュベートしてください。
- 各免疫沈降ごとにチューブをMagnetic Separation Rackにセットし、Protein G Magnetic Beadsを分離させてください。溶液が澄むまで1-2分間待ち、上清を注意深く取り除いてください。直ちにセクションVIに進んでください。
VI. 抗体/Protein G Magnetic Beadsからのクロマチンの溶出および脱クロスリンク
実験開始前の準備:
- 37℃のウォーターバスで2X ChIP Elution Buffer #7009を温め、SDSが完全に溶解していることを確認してください。
- 水浴またはサーモミキサーを65℃に設定してください。
- それぞれの免疫沈降につき、1X ChIP Elution Buffer 150 µL (2X ChIP Elution Buffer #7009 75 µL + 水 75 µL) と2%インプットサンプルを調製してください。
- 1X ChIP Elution Buffer 150 μLを、2%インプットサンプル入りのチューブに加え、ステップ7で使用するまで室温に置いてください。
- 1X ChIP Elution Buffer 150 μLを、それぞれの免疫沈降サンプルに加えてください。
- 静かにボルテックス (1,200 rpm) しながら、65℃で30分間処理し、抗体/Protein G Magnetic Beads複合体からクロマチンを溶出してください。本ステップには、サーモミキサーの使用を推奨しますが、頻繁に攪拌しながら65°Cのウォーターバスを使用することも可能です。代わりにローテーターを用いて室温で溶出することもできますが、完全に溶出されない可能性があります。
- サンプルを、10,000 x gで10秒間遠心し、チューブ蓋に付着したサンプルを除いてください。
- チューブをMagnetic Separation Rackにセットし、液が澄むまで1-2分間待ち、Protein G Magnetic Beadsを分離させてください。
- 溶出されたクロマチンを含む上清を、注意深く新しいチューブに移してください。
- ステップ1で調製した2%インプットサンプルを含むすべてのチューブに、5M NaCl #7010 6 μLおよび#10012 Proteinase K 2 μLを加え、65℃で2時間インキュベートし、脱クロスリンクしてください。インキュベーションは一晩まで延長することができます。
- 直ちにセクションVIIに進んでください。もしくは、サンプルを‐20℃で保存してください。ただし、沈殿物の形成を防ぐために、DNA Binding Buffer #10007を加える前に (セクションVIIのステップ1)、必ずサンプルを室温に戻してください。
VII. スピンカラムを用いたDNAの精製
実験開始前の準備:
- 使用前に、DNA Wash Buffer #10008にエタノール (96 - 100%) 24 mLを加えてください。このステップは、DNA精製の最初のセッティングの前に1度だけ実施してください。
- セクションIVで得られたDNAサンプルそれぞれに対して、DNA精製用スピンカラムとコレクションチューブ#10010を一つずつ用意してください。
- DNA Binding Buffer #10007 750 μLを各DNAサンプルに加え、軽くボルテックスしてください。
- 1サンプルあたり5倍量のDNA Binding Bufferを使用します。
- ステップ1の各DNAサンプル450 μLを、コレクションチューブにセットしたDNAスピンカラムに移してください。
- 14,000 rpmで30秒間遠心分離してください。
- コレクションチューブからスピンカラムを取り外し、液体を廃棄してください。スピンカラムをコレクションチューブに再セットしてください。
- ステップ1で残った各DNAサンプル450 μLを、コレクションチューブにセットしたスピンカラムに移してください。ステップ3と4を繰り返してください。
- DNA Wash Buffer #10008 750 μLをコレクションチューブにセットしたスピンカラムに加えてください。
- 14,000 rpmで30秒間遠心分離してください。
- コレクションチューブからスピンカラムを取り外し、液体を廃棄してください。スピンカラムをコレクションチューブに再セットしてください。
- 14,000 rpmで30秒間遠心分離してください。
- コレクションチューブと液体を廃棄してください。スピンカラムは廃棄しないでください。
- DNA Elution Buffer #10009 50 μLを各スピンカラムに加え、新しい1.5 mLの遠心分離用チューブにセットしてください。
- 14,000 rpmで30秒間遠心分離し、DNAを溶出してください。
- DNAスピンカラムを取り外し、廃棄してください。得られた溶出液が精製されたDNAです。サンプルは-20℃で保存することができます。
VIII. PCRによるDNAの定量
推奨事項:
- コンタミネーションを防ぐため、フィルターチップ付きピペットを使用してください。
- キットに含まれているコントロールプライマーは、ヒトとマウスのRPL30遺伝子 (#7014と#7015) に特異的で、通常のPCRまたは定量的リアルタイムPCRに対して使用することができます。他の動物種でChIPを実施する場合は、その種で使用可能な適切なプライマーを設計し、最適なPCR条件を決定してください。
- 非特異的なPCR産物の増幅を防ぐため、ホットスタート用Taqポリメラーゼの使用を推奨します。
- プライマーの設定は非常に重要です。以下の条件に近いプライマーを設計してください:
| プライマーの長さ: |
24塩基 |
| Tm: |
60°C |
| GC: |
50% |
| アンプリコンのサイズ: |
150-200 bp (通常のPCR) |
|
80-160 bp (定量的リアルタイムPCR) |
通常のPCR:
- 使用するPCR装置のモデルに対応するPCRチューブあるいはPCRプレートに、適切なサンプル番号を記載してください。2%インプットサンプル、ポジティブコントロールのHistone H3サンプル、ネガティブコントロールのNormal Rabbit IgGサンプル、およびDNAコンタミネーションに対するコントロールとしてDNAなしのチューブを用意してください。
- 各チューブにDNAサンプル2 μLを分注してください。
- 分量不足を防ぐために、実際のチューブの本数に2本追加した量を全体量として、マスターミックスを下記の通りに調製してください。各反応用チューブにマスターミックス18 μLを加えてください。
| 試薬 |
PCR反応1回分の分量 (18 μL) |
| Nuclease-free H2O |
12.5 µL |
| 10X PCR Buffer |
2.0 µL |
| 4 mM dNTP Mix |
1.0 µL |
| 5 µM RPL30 プライマー |
2.0 µL |
| Taq DNA Polymerase |
0.5 µL |
- PCR反応を以下のプログラムで開始してください:
- 初期変性 95℃ 5分間
- 変性 95℃ 30秒間
- アニーリング 62℃ 30秒間
- 伸長反応 72℃ 30秒間
- ステップb-dを繰り返し、計34サイクル反応させてください。
- 最終伸長反応 72℃ 5分間
- 反応終了後、100 bpのDNAマーカーとともに、各PCR産物10 μLを2%アガロースゲルまたは10%ポリアクリルアミドゲルで電気泳動してください。予想されるPCR産物のサイズは、Human RPL30 #7014では161 bp、Mouse RPL30 #7015 では159 bpです。
定量的リアルタイムPCR:
- 使用するPCR装置のモデルに対応するPCRチューブあるいはPCRプレートに、適切なサンプル番号を記載してください。PCR反応には、ポジティブコントロールのHistone H3サンプル、ネガティブコントロールのNormal Rabbit IgGサンプル、DNAコンタミネーション確認用のコントロールとしてDNAを含まないチューブ、および標準曲線の作成と増幅効率の決定のため2%インプットクロマチンDNAの段階希釈サンプル (希釈なし、1:5、1:25、1:125) を用意してください。
- PCRチューブあるいはPCRプレートのウェルに、適切なDNAサンプル2 μLを加えてください。
- 以下のように、マスターミックスを調製してください。PCR反応ごとに2-3の複製サンプルをセットアップしてください。分量不足を考慮して、マスターミックスは余裕を持った量で調製してください。各PCRチューブあるいは各ウェルにマスターミックス18 μLを加えてください。PCR反応一回分の試薬量 (18 µL)
| Nuclease-free H2O |
6 µL |
| 5 µM RPL30 プライマー |
2 µL |
| SimpleChIP® Universal qPCR Master Mix #88989 |
10 µL |
- PCR反応を以下のプログラムで開始してください:
| a. |
初期変性 |
95℃ 3分間 |
| b. |
変性 |
95℃ 15秒間 |
| c. |
アニーリングおよび伸長: |
60℃ 60秒間 |
| d. |
ステップb - cの繰り返し (合計40サイクル) |
|
- リアルタイムPCR装置に付属のソフトウェアを使用して、定量結果を解析してください。代替法として、Percent Input法により下記の公式を用いて、免疫沈降の効率を算出することもできます。この方法では、各免疫沈降で回収されたシグナルを、インプットクロマチン総量の割合 (%) で示します。
Percent Input = 2% x 2(C[T] 2%Input Sample - C[T] IP Sample)
C[T] = CT = Average threshold cycle of PCR reaction
IX. NG-seqライブラリーの調製
本キットを用いて調製したDNAサンプルは、直接ChIP-seqに使用できます。NG-シークエンスDNAライブラリーの構築は、採用予定のシーケンスプラットフォームと互換性のあるDNAライブラリー調製プロトコールまたはキットを使用して行ってください。Illuminaプラットフォームでのシーケンシングの場合、DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795と、関連するインデックスプライマーMultiplex Oligos for Illumina (Single Index Primers) (ChIP-seq, CUT&RUN) #29580もしくはMultiplex Oligos for Illumina (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538の使用を推奨します。
推奨事項:
- 転写因子またはコファクターのChIP-seqでは、ChIPで濃縮したDNAを少なくとも5 ng使用し、10サイクルのPCRでアダプター配列が結合したDNAを増幅してください。
- Totalヒストン、ヒストン修飾、そしてインプットサンプルについては、ChIPで濃縮したDNA 50 ngを使用するところから始め、6サイクルのPCRでアダプター配列が結合したDNAを増幅してください。
- どんなターゲットの種類に対しても、ChIPで濃縮したDNAのライブラリー構築でサイズセレクションを行わずに、アダプター配列が結合したDNAをクリーンナップしてください。
- DNAライブラリー構築後、Agilent High Sensitivity DNA Kit (Agilent Technologies, Cat# G2938-90322)、または2%アガロースTAEゲルを用いて50-100 ngのDNAを電気泳動し、アダプターダイマー (約140 bp) がDNAライブラリーに存在するかどうかを確認してください。アダプターダイマーが存在する場合、PCR増幅資材の洗浄を繰り返します。
- ライブラリーの品質は、既知のポジティブおよびネガティブコントロール用プライマーセットを用いたリアルタイムPCRにより確認することもできます。オリジナルのChIP産物を用いた結果と同様に、DNAライブラリーでもポジティブコントロールはネガティブコントロールと比較して高いシグナルが得られるはずです。
- 最終的なクリーンアップおよび品質チェックの後、最終精製ライブラリーサンプルを2 - 10 nMに調製してハイスループットシーケンシングに使用してください。
Appendix A:予想されるクロマチン収量
組織サンプルからクロスリンククロマチンを調製する場合、組織の種類によってクロマチン収量が大きく異なります。下記テーブルに、各組織100 mgまたはHCT 116細胞2 x 107個から得られると予想されるクロマチン収量と予想されるDNA濃度 (Section IVのプロトコールで測定) を記載しました。ChIPの最適な結果を得るためには、 DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795と、関連するインデックスプライマーMultiplex Oligos for Illumina (Single Index Primers) (ChIP-seq, CUT&RUN) #29580もしくはMultiplex Oligos for Illumina (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538の使用を推奨します。
| 組織/細胞 |
総クロマチン収量 |
予想されるDNA濃度 |
| 肝臓 |
100 mgの組織に対し50 μg |
150 µg/mL |
| 脳 |
100 mgの組織に対し25 μg |
50 µg/mL |
| 心臓 |
100 mgの組織に対し105 μg |
20 µg/mL |
| HCT 116 |
2 x 107個の細胞に対し100-150 µg |
100 - 150 µg/mL |
Appendix B:ホルムアルデヒドによる固定条件の最適化
転写因子や転写コファクターは、ヒストンタンパク質よりも緩やかにクロマチンDNAに結合します。そのため、ソニケーション処理中にクロマチンから分離する傾向にあります。特に、組織サンプルの場合、固定時間を長くすることで、ChIPアッセイにおいて転写因子や転写コファクターが補足される量を増やすことができます。図7に示すように、固定時間を10分間から30分間に延ばすことで、クロマチン断片化の効率が低下することがありますが (左パネル)、転写コファクターであるRING1BやSUZ12の濃縮効率は向上することが、心臓組織のChIP-qPCRで示されています (中央および右パネル)。
一般的に、ヒストン修飾のChIPでは、細胞でも組織でも10分間の固定で十分ですが、転写因子や転写コファクターでは、特に組織サンプルの場合、30分間以上の固定を必要とします。
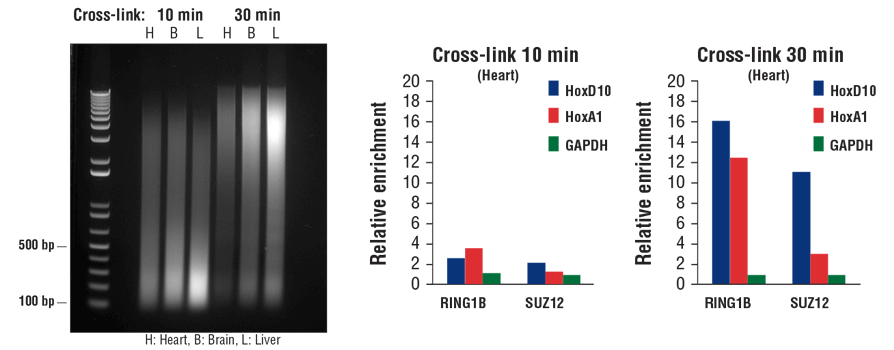
図7 マウスの心臓 (H)、脳 (B)、および肝臓 (L) を10分間または30分間クロスリンクしました (左パネル)。調製したクロマチンはソニケーション処理し、精製したDNA20 μLを1%アガロースゲルで電気泳動して分離しました。ChIP-qPCRアッセイ (中央と右パネル) では、RING1B (D22F2) XP® Rabbit mAb #5694 10 µLあるいは、SUZ12 (D39F6) XP® Rabbit mAb #3737 5 µLのどちらかを用いてクロマチン免疫沈降を実施しました。濃縮したDNAは、SimpleChIP® Mouse HoxD10 Exon 1 Primers #7429、SimpleChIP® Mouse HoxA1 Promoter Primers #7341、およびSimpleChIP® Mouse GAPDH Intron 2 Primers #8986を用いて、リアルタイムPCRにより解析しました。ネガティブコントロールのGAPDHのシグナルを1とした場合の相対値として、各サンプルのシグナルを示しています。
Appendix C: クロマチン断片化の最適化
クロスリンククロマチンDNAの断片化についての最適条件は、細胞数、サンプル量、ソニケーション時間、使用するソニケーターパワーの設定により大きく異なります。各ソニケーションサンプルに対し、100-150mgの組織または1x107-2x107個の細胞を、1mLの ChIP Sonication Nuclear Lysis Bufferを使用することを推奨しています。ご使用になる組織や細胞タイプで最適なソニケーション条件を決定するためのプロトコールを以下に示します。
- セクションI、II、IIIに従い、100-150mgの組織または1x107-2x107個の細胞を用いてクロスリンクした細胞核を調製してください。セクションIIIのステップ4を行った後、下記に進んでください。
- ソニケーションによるクロマチンの断片化:最適な超音波処理条件はソニケーターごとに異なり、出力設定に応じて超音波処理の回数と持続時間を調節する必要があります (Branson Digital Sonifier 250 probe sonicatorの推奨出力設定については、セクションIIIのステップ5をご参照ください)。最適なソニケーション条件を決定するには、ソニケーションのタイムコースを設定し、一定の回数または時間ソニケーションした後、50 μLのクロマチンサンプルを採取してください。例えば、ソニケーション処理をしながら1 - 2分おきに、クロマチンサンプルを採取してください。
- 21,000 x g、4℃で10分間遠心分離し、クロマチンサンプルを清澄化してください。
- 上清を新しい遠心分離用チューブに移し、Nuclease-free water 100 μL、5M NaCl #7010 6 μL、RNAse A #7013 2 μLを加えてください。ボルテックスで混和し、37℃で30分間インキュベートしてください。
- 各サンプルに、Proteinase K #10012 2 μLを加えます。ボルテックスにより混和し、65℃で2時間インキュベートしてください。
- 各サンプルから20 µLを取り出し、DNA断片のサイズを調べてください。
- 最適なDNA断片サイズが得られるソニケーション条件を選択して、セクションIIIのステップ5のクロマチン調製に用いてください。最適なソニケーション条件が得られない場合は、ソニケーターの出力設定を増減してソニケーションのタイムコースの検討を繰り返してください。
注意:最適なソニケーション条件はサンプルの種類や固定時間によって異なります。推奨されるサイズのクロマチン断片を得るために必要な、最小回数のソニケーションを行うようにしてください。長さが500 bp未満のDNA断片が全体の80%を超えるような過剰なソニケーションを行うと、クロマチンに過度のダメージを与え、免疫沈降の効率が低下する恐れがあります (図8、右のパネル参照)。
- 10分間固定した細胞の場合、最適なソニケーション条件下では、DNA断片全体の約90%が1 kb未満のスメア状のバンドとして観察されます (図8、左のパネル参照)。固定時間を30分に延長すると断片化の効率が低下し、1 kb未満のスメア状のバンドとして観察されるDNA断片は約60%となります。
- 10分間固定した組織の場合、最適なソニケーション条件下では、DNA断片全体の約60%が1 kb未満のスメア状のバンドとして観察されます。固定時間を30分に延長すると断片化の効率が低下し、1kb未満のスメア状のバンドとして観察されるDNA断片は約30%となります (図7、左のパネルを参照)。
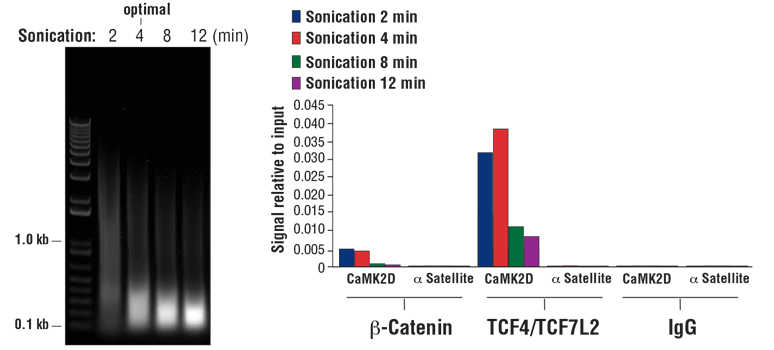
図8 クロマチン免疫沈降は、10分間クロスリンクし指定時間ソニケーション処理した2x107 HCT 116細胞を用いて実施しました。#56383SimpleChIP® Plus Sonication Chromatin IP KitのセクションIV記載の方法で精製し、20 µLの精製したDNAを1%アガロースゲルの電気泳動により分析しました。ソニケーションサイクルを増やすと、クロマチン断片のサイズが小さくなりました (左パネル)。クロマチン免疫沈降は、#56383 SimpleChIP® Plus Sonication Chromatin IP Kitと、#8814 Non-phospho (Active) Β-Catenin (Ser33/37/Thr41) (D13A1) Rabbit mAb 5µL、#2569 TCF4/TCF7L2 (C48H11) Rabbit mAb 10 µL、#2729 Normal Rabbit IgG 2 µLを用いて行いました。SimpleChIP® Human CaMK2D Intron 3 Primers #5111、およびSimpleChIP® Human α Satellite Repeat Primers #4486を用いたリアルタイムPCRにより、免疫沈降したDNAを解析しました。各サンプルにおける免疫沈降されたDNA量は、インプットクロマチンの総量を1とした場合の相対値として表示しました (右パネル)。この結果から、Branson Digital Sonifier D250プローブソニケーターの1/8インチのマイクロチップを使用した場合、4分間のクロマチンソニケーションで最適の結果が得られることが分かります。過剰なソニケーション処理によって、コファクターのbeta-Cateninと転写因子のTCF4/TCF7L2に対する抗体を用いたクロマチンの濃縮効率が著しく低下しました。
Appendix D:トラブルシューティングガイド
| 問題 |
考えられる原因 |
推奨される対処法 |
| 1. 断片化DNAの濃度が低すぎる。 |
細胞/核の溶解が不完全である。
クロマチン調製に十分な細胞数を使用していない。
|
クロマチンサンプルのDNA濃度が50 μg/mLに近い場合は、1回の免疫沈降あたり少なくとも5 μgとなるようにクロマチン調製液を追加して、プロトコールに従って実験を続けてください。
クロスリンクの前に、別途用意したプレートで細胞数を数え、正確な細胞数を測定してください。
|
クロスリンク時間を10-30分間の範囲に短縮してください。ソニケーション1回あたりに使用する細胞/組織の量を減らしてください。ソニケーションのタイムコース実験を行ってください。
| 2. クロマチンの断片化が不十分で、DNA断片が大きすぎる (>50% 1.5 kb以上)。 |
細胞のクロスリンクが過剰。
用いる細胞/組織量が多すぎる。
|
クロスリンク時間を10-30分間の範囲に短縮してください。ソニケーション1回あたりに使用する細胞/組織の量を減らしてください。ソニケーションのタイムコース実験を行ってください。
|
| 3. クロマチンの断片化が進行しすぎている (>90% 300 bp未満)。 |
ソニケーション処理条件が厳しすぎる。
|
ソニケーションのタイムコース実験を行い、適切な断片サイズを得るための最低限の処理時間を確認してください。
|
| 4. インプットDNAのPCR反応で産物が得られない、または得られる量が非常に少ない。 |
PCR反応に使用したDNA量が十分でない、またはPCR条件が最適でない。
PCRの増幅領域が、ヌクレオソームフリー領域に及んでいる。
IPに加えたクロマチン量が十分でない、またはソニケーション処理が過剰。
|
PCR反応により多くのDNAを使用するか、サイクル数を増加させてください。
クロスリンク・ソニケーション処理したクロマチンから精製したDNAを用いて、PCR条件をプライマーに対して最適化してください。最適なChIP結果を得るために、免疫沈降1回あたりクロマチン5-10 μgを加えてください。上記1、3の対応策も参照してください。
|
| 5. ポジティブコントロールであるHistone H3抗体の免疫沈降サンプルとRPL30プライマーを用いたPCR反応で増幅が起こらない。 |
免疫沈降に加えたクロマチンまたは抗体が十分でない、または免疫沈降のインキュベーション時間が短すぎる。
Protein G Beadsからのクロマチンの溶出が不十分である。
|
各免疫沈降反応に、少なくともクロマチン5-10 μgと抗体10 μLを加えて一晩インキュベートし、Protein Gビーズを加え、さらに2時間インキュベートしてください。
Protein G Beadsからのクロマチンの溶出には65℃が最適です。頻繁に撹拌してBeadsの懸濁状態を保ってください。
|
| 6. ネガティブコントロールのRabbit IgGの免疫沈降産物と、ポジティブコントロールのHistone H3抗体の免疫沈降産物で、PCRでの増幅が同程度になる。 |
免疫沈降に使用したクロマチン量が過剰、または十分でない。あるいは、免疫沈降に抗体を加えすぎている。
PCR反応に加えたDNAが多すぎる、またはサイクル数が多すぎる。
|
各免疫沈降反応に対して加える量は、クロマチン15 μg、Histone H3 antibody 10 μLを上限としてください。Normal rabbit IgG量を一回のIPあたり1 μLまで減らしてください。
PCR反応に加えるDNAを減らすか、PCRのサイクル数を減らしてください。PCRの線形増幅領域内でPCR産物を解析することが非常に重要です。そうしなければ、増幅前のDNA量の差が正確に測定できません。
|
| 7. 解析したいターゲットの抗体の免疫沈降産物で、PCR反応による増幅が起こらない。 |
PCR反応に加えたDNA量が十分でない。
免疫沈降に使用した抗体量が十分でない。
免疫沈降ではワークしない抗体である。
|
PCR反応により多くのDNAを使用するか、サイクル数を増加させてください。
通常は免疫沈降反応液に1-5 μgの抗体を加えますが、実際に必要な量は抗体によって大きく異なります。
免疫沈降反応液に加える抗体の量を増やしてください。別の抗体を検討してください。
|


