View in English?
View in English?
View in English?

| Cat. # | Size | Qty. | Price | Inventory |
|---|---|---|---|---|
| 3580S | 100 µl |
|
| REACTIVITY | H |
| SENSITIVITY | Endogenous |
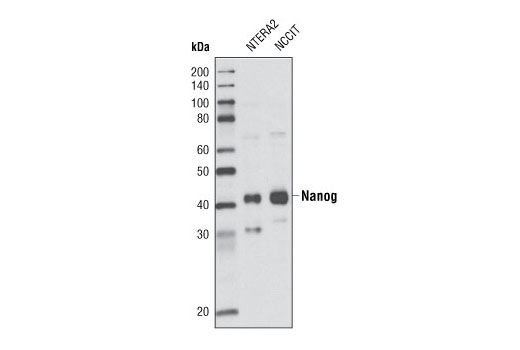
| MW (kDa) | 42 |
| SOURCE | Rabbit |
Product Information
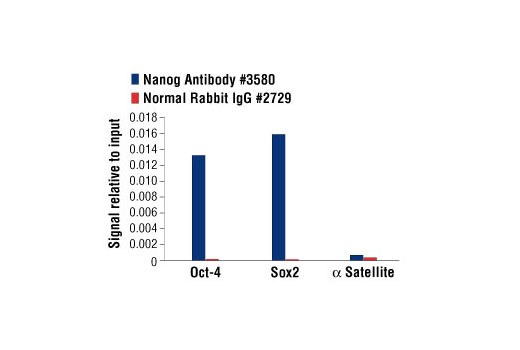
For optimal ChIP results, use 20 μl of antibody and 10 μg of chromatin (approximately 4 x 106 cells) per IP. This antibody has been validated using SimpleChIP® Enzymatic Chromatin IP Kits.
| Application | Dilution |
|---|---|
| Western Blotting | 1:1000 |
| Immunofluorescence (Immunocytochemistry) | 1:800 |
| Flow Cytometry (Fixed/Permeabilized) | 1:400 |
| Chromatin IP | 1:25 |
For western blots, incubate membrane with diluted primary antibody in 5% w/v BSA, 1X TBS, 0.1% Tween® 20 at 4°C with gentle shaking, overnight.
NOTE: Please refer to primary antibody product webpage for recommended antibody dilution.
From sample preparation to detection, the reagents you need for your Western Blot are now in one convenient kit: #12957 Western Blotting Application Solutions Kit
NOTE: Prepare solutions with reverse osmosis deionized (RODI) or equivalent grade water.
Load 20 µl onto SDS-PAGE gel (10 cm x 10 cm).
NOTE: Loading of prestained molecular weight markers (#59329, 10 µl/lane) to verify electrotransfer and biotinylated protein ladder (#7727, 10 µl/lane) to determine molecular weights are recommended.
NOTE: Volumes are for 10 cm x 10 cm (100 cm2) of membrane; for different sized membranes, adjust volumes accordingly.
* Avoid repeated exposure to skin.
posted June 2005
revised June 2020
Protocol Id: 10
Achieve higher quality immunofluorescent images using the efficient and cost-effective, pre-made reagents in our #12727 Immunofluorescence Application Solutions Kit
NOTE: Prepare solutions with reverse osmosis deionized (RODI) or equivalent grade water.
Recommended Fluorochrome-conjugated Anti-Rabbit secondary antibodies:
NOTE: Cells should be grown, treated, fixed and stained directly in multi-well plates, chamber slides or on coverslips.
Aspirate liquid, then cover cells to a depth of 2–3 mm with 4% formaldehyde diluted in 1X PBS.
NOTE: Formaldehyde is toxic, use only in a fume hood.
NOTE: All subsequent incubations should be carried out at room temperature unless otherwise noted in a humid light-tight box or covered dish/plate to prevent drying and fluorochrome fading.
posted November 2006
revised November 2013
Protocol Id: 24
All reagents required for this protocol may be efficiently purchased together in our Intracellular Flow Cytometry Kit (Methanol) #13593, or individually using the catalog numbers listed below.
NOTE: Prepare solutions with reverse osmosis deionized (RODI) or equivalent grade water.
NOTE: When including fluorescent cellular dyes in your experiment (including viability dyes, DNA dyes, etc.), please refer to the dye product page for the recommended protocol. Visit www.cellsignal.com for a full listing of cellular dyes validated for use in flow cytometry.
NOTE: Adherent cells or tissue should be dissociated and in single-cell suspension prior to fixation.
NOTE: Optimal centrifugation conditions will vary depending upon cell type and reagent volume. Generally, 150-300g for 1-5 minutes will be sufficient to pellet the cells.
NOTE: If using whole blood, lyse red blood cells and wash by centrifugation prior to fixation.
NOTE: Antibodies targeting CD markers or other extracellular proteins may be added prior to fixation if the epitope is disrupted by formaldehyde and/or methanol. The antibodies will remain bound to the target of interest during the fixation and permeabilization process. However, note that some fluorophores (including PE and APC) are damaged by methanol and thus should not be added prior to permeabilization. Conduct a small-scale experiment if you are unsure.
NOTE: Count cells using a hemocytometer or alternative method.
posted July 2009
revised June 2020
Protocol Id: 404
SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) #9005の専用プロトコールです。
キットに含まれている試薬:
キットに含まれない試薬:
| ! | この ! マークは、免疫沈降 (IP prep) のサンプル数に応じて量の変更する重要ステップであることを意味します。免疫沈降1回分 (1 IP prep) は、4 x 106個の組織培養細胞、または25 mgの破砕した組織に相当します。 |
| !! | この !! マークは、操作を進める前にバッファーを希釈する重要なステップであることを意味します。 |
| SAFE STOP | これは、実験操作を中断する必要がある場合に、プロトコールを安全に中断できるポイントを示します。 |
組織を採取する際、脂肪や壊死組織などの不要な部分を取り除きます。組織サンプルはすぐに使用して直ちにクロスリンクするか、次に使用するまでドライアイスで凍結して-80℃で保存してください。最適なクロマチン収量とChIP結果を得るため、各免疫沈降あたり25 mgの組織を使用します。クロマチン収量は組織の種類により異なるため、一部の組織では各免疫沈降あたり25 mgを超える量が必要となります。組織ごとに予測されるクロマチン収量については、Appendix Aを参照してください。クロマチンの断片化および濃度の解析 (セクションIV) のためにクロマチンサンプルを1本余分に用意してください。必要に応じて5本のクロマチンサンプルを追加で用意し、クロマチン断片化の最適化 (Appendix B) で使用します。
(!) すべてのバッファーの量は、免疫沈降 (IP prep) のサンプル数に応じて比例的に増やす必要があります。
最適なChIPの結果を得るため、各免疫沈降に対し約4 x 106個の細胞を使用します。ポジティブコントロールとネガティブコントロールを含めると、少なくとも12 x 106個の細胞が必要です。HeLa細胞の場合、免疫沈降1回分は増殖培地20 mLで90%コンフルエントに培養した15 cmディッシュの半分量に相当します。クロマチンサンプルを1本余分に用意し、クロマチンの断片化および濃度についての解析に使用します (セクションIV)。さらに、細胞種ごとに培養ディッシュを1枚余分に用意し、血球計算盤またはセルカウンターを用いて細胞数を計測することを推奨します。
(!) 全てのバッファーの量は、使用する15 cmの組織培養ディッシュ (または20 mLの浮遊細胞) の枚数に応じて、比例的に増やす必要があります。
(!) すべてのバッファーの量は、免疫沈降 (IP prep) のサンプル数に応じて比例的に増やす必要があります。
(!!) 重要:溶解したら、1M DTTは-20℃で保存してください。
注意:最適なChIPの結果を得るためには、クロマチンのサイズと濃度を適切にすることが非常に重要です。クロマチンを断片化しすぎると、定量PCRのシグナルが消失する可能性があります。一方で、クロマチンの断片化が不十分な場合、バックグラウンドシグナルが上昇し分解能が低下します。また、免疫沈降に用いるクロマチン量が少なすぎると、定量PCRのシグナルが低下する原因になります。クロマチンの断片化を最適化するためのプロトコルは、Appendix Bに記載されています。
最適なChIPの結果を得るため、免疫沈降1回あたり約5-10 μgの断片化されたクロスリンククロマチン (セクションIVに記載) を使用してください。これは、25 mgの破砕した組織あるいは4 x 106個の培養細胞から調製された免疫沈降サンプル100 μL分に概ね相当します。一般的には、抗体を加える前にクロマチンサンプル100 μLを1X ChIP Buffer 400 μLで希釈しますが、免疫沈降1回あたり100 μL以上必要な場合は下記のようにクロマチンサンプルを希釈する必要はありません。希釈しない場合も、クロマチンサンプルに直接抗体を加えることでクロマチン複合体の免疫沈降ができます。
(!) すべてのバッファーの量は、免疫沈降の回数に応じて比例的に増やす必要があります。
注意:Cell Signaling Technologyのほとんどの抗体は、免疫沈降1回あたり1-2 µgを使用することで適切にワークします。濃度の異なる複数の抗体がある場合は、最も高い抗体濃度にネガティブコントロールNormal Rabbit IgG #2729の濃度を合わせて使用することをお勧めします。
(!) すべてのバッファーの量は、免疫沈降の回数に応じて比例的に増やす必要があります。
| プライマーの長さ: | 24塩基 |
| Tm: | 60°C |
| GC: | 50% |
| アンプリコンのサイズ: | 150-200 bp (通常のPCR) |
| 80-160 bp (定量的リアルタイムPCR) |
| 試薬 | PCR反応1回分の分量 (18 μL) |
|---|---|
| Nuclease-free H2O | 12.5 µL |
| 10X PCR Buffer | 2.0 µL |
| 4 mM dNTP Mix | 1.0 µL |
| 5 µM RPL30 プライマー | 2.0 µL |
| Taq DNA Polymerase | 0.5 µL |
| a. | 初期変性 | 95℃ | 5分間 |
| b. | 変性 | 95℃ | 30 秒間 |
| c. | アニーリング | 62℃ | 30 秒間 |
| d. | 伸長反応 | 72℃ | 30 秒間 |
| e. | ステップb-dを繰り返し、計34サイクル反応させてください。 | ||
| f. | 最終伸長反応 | 72℃ | 5分間 |
| 試薬 | PCR反応1回分の分量 (18 μL) |
|---|---|
| Nuclease-free H2O | 6 µL |
| 5 µM RPL30 プライマー | 2 µL |
| SimpleChIP® Universal qPCR Master Mix #88989 | 10 µL |
| a. | 初期変性 | 95℃ 3分間 |
| b. | 変性 | 95℃ 15秒間 |
| c. | アニーリングおよび伸長: | 60℃ 60秒間 |
| d. | ステップb - cの繰り返し (合計40サイクル) | |
リアルタイムPCR装置に付属のソフトウェアを使用して、定量結果を解析してください。代替法として、Percent Input法により下記の公式を用いて、免疫沈降の効率を算出することもできます。この方法では、各免疫沈降で回収されたシグナルを、インプットクロマチン総量の割合 (%) で示します。
Percent Input = 2% x 2(C[T] 2%Input Sample - C[T] IP Sample)
C[T] = CT= PCR反応の閾値となるサイクル
本キットを用いて調製したDNAサンプルは、直接ChIP-seqに使用できます。NG-シークエンスDNAライブラリーの構築は、採用予定のシーケンスプラットフォームと互換性のあるDNAライブラリー調製プロトコールまたはキットを使用して行ってください。Illumina®プラットフォームでのシーケンシングの場合、DNA Library Prep Kit for Illumina® (ChIP-seq, CUT&RUN) #56795と、関連するインデックスプライマーMultiplex Oligos for Illumina® (Single Index Primers) (ChIP-seq, CUT&RUN) #29580もしくは、Multiplex Oligos for Illumina® (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538の使用を推奨します。
推奨事項:
組織サンプルからクロスリンククロマチンを調製する場合、組織の種類によってクロマチン収量が大きく異なります。右の表には、4 x 106個のHeLa細胞の場合と比較しながら、25 mgの組織から調製した場合に予想されるクロマチン収量と、予想されるDNA濃度を示しています。これらは、プロトコールのセクションIVに記載した方法で決定しています。各組織において、Medimachine (BD Biosciences) またはDounce Homogenizerのどちらを用いてホモジナイズした場合もクロマチン収量はほぼ同じでした。ただし、多くの場合、Medimachineを用いて破砕した組織から得たクロマチンは、Dounce Homogenizerを用いた場合よりも免疫沈降効率が高くなりました。脳組織サンプルに関しては、Medimachineでは単細胞懸濁液になるまで十分に破砕することができないため、Dounce Homogenizerを使用してください。最適なChIPの結果を得るためには、免疫沈降1回あたり5-10 µgの断片化したクロスリンククロマチンの使用をお勧めします。組織によっては、免疫沈降1回に必要な組織量が25 mgを超えることがあります。
| 組織/細胞 | 総クロマチン収量 | 予想されるDNA濃度 |
|---|---|---|
| 脾臓 | 25 mgの組織に対し20-30 µg | 200 - 300 µg/mL |
| 肝臓 | 25 mgの組織に対し10-15 µg | 100 - 150 µg/mL |
| 腎臓 | 25 mgの組織に対し8-10 µg | 80 - 100 µg/mL |
| 脳 | 25 mgの組織に対し2-5 µg | 20 - 50 µg/mL |
| 心臓 | 25 mgの組織に対し2-5 µg | 20 - 50 µg/mL |
| HeLa | 4 x 106個の細胞に対し10-15 µg | 100 - 150 µg/mL |
クロスリンクしたクロマチンDNAを150-900 bpの長さに断片化する場合の最適条件は、断片化に用いる組織量または細胞数に対するMicrococcal Nucleaseの比率に大きく依存します。特定の組織または細胞タイプで、クロマチンの断片化の至適条件を決定するプロトコールを以下に示します。
| 問題 | 考えられる原因 | 推奨される対処法 |
|---|---|---|
| 1. 断片化されたクロマチンの濃度が低すぎる。 | 細胞数が不十分、あるいは断片化後の細胞核の破砕が不完全。 | クロマチンサンプルのDNA濃度が50 μg/mLに近い場合は、1回の免疫沈降あたり少なくとも5 μgとなるようにクロマチン調製液を追加して、プロトコールに従って実験を続けてください。 クロスリンクの前に、カウント用に別に用意したディッシュで細胞数を計測し、正確な細胞数を確認してください。さらに、顕微鏡下でソニケーション前後の細胞核を観察し、核の完全な破砕を確認してください。 |
| 2. クロマチンの断片化が不十分で、断片が大きすぎる (900 bpを超える)。 | 細胞のクロスリンクが過剰。10分間以上クロスリンクを行うと、クロマチンの断片化を阻害する可能性がある。 細胞数が多すぎる、またはMicrococcal Nuclease量が足りない。 | ホルムアルデヒド濃度を一定にして、時間経過に伴う変化を確認してください。クロスリンク時間を10分以下に短縮してください。 クロスリンクの前に別に用意したプレートの細胞数を計測し、Appendix Bを参照してクロマチンの断片化条件を最適化します。 |
| 3. クロマチンが過剰に断片化され、DNA断片のサイズが小さすぎる (150 bpのモノヌクレオソーム1個分の断片ばかりが検出される)。ヌクレオソーム1個分のDNAの長さになるまでクロマチンを完全に断片化すると、特に長さが150 bp以上のアンプリコンの場合、PCRでシグナルが減弱する可能性がある。 | クロマチン断片化に加えた細胞数が不十分、またはMicrococcal Nuclease量が多すぎる。 | クロスリンクの前に別に用意したプレートの細胞数を計測し、Appendix Bを参照してクロマチンの断片化条件を最適化します。 |
| 4. インプットDNAのPCR反応で産物が得られない、または得られる量が非常に少ない。 | PCR反応に使用したDNA量が十分でない、またはPCR条件が最適でない。 PCRの増幅領域が、ヌクレオソームフリー領域に及んでいる。 IPに加えたクロマチンが十分でない、またはクロマチンの断片化が過剰。 | PCR反応により多くのDNAを使用するか、サイクル数を増加させてください。 クロスリンク後に断片化したクロマチンから精製したDNAを用いて、プライマーセットに対する最適なPCR条件を検討してください。アンプリコンの長さが150 bp以下になるように、別のプライマーセットを設計してください (セクションIIIのプライマー設計に関する推奨事項を参照してください)。 最適なChIP結果を得るために、免疫沈降1回あたりクロマチン5-10 μgを加えてください。上記1、3の対応策も参照してください。 |
| 5. ポジティブコントロールであるHistone H3抗体の免疫沈降サンプルとRPL30プライマーを用いたPCR反応で増幅が起こらない。 | 免疫沈降に加えたクロマチンまたは抗体が十分でない、または免疫沈降のインキュベーション時間が短すぎる。 Protein G Beadsからのクロマチンの溶出が不十分である。 | 各免疫沈降反応にクロマチン5-10 μgと抗体10 μLを加えたことを確認して一晩インキュベートし、Protein G Beadsを加えてさらに2時間インキュベートしてください Protein G Beadsからのクロマチンの溶出には65℃が最適です。頻繁に撹拌してBeadsの懸濁状態を保ってください。 |
| 6. ネガティブコントロールのRabbit IgGの免疫沈降産物と、ポジティブコントロールのHistone H3抗体の免疫沈降産物で、PCRでの増幅が同程度になる。 | 免疫沈降に使用したクロマチン量が過剰、または十分でない。あるいは、免疫沈降に抗体を加えすぎている。 PCR反応に加えたDNAが多すぎる、またはサイクル数が多すぎる。 | 各免疫沈降反応に対して加える量は、クロマチン15 μg、Histone H3 antibody 10 μLを上限としてください。Normal rabbit IgG量を一回のIPあたり1 μLまで減らしてください。 PCR反応に加えるDNAを減らすか、PCRのサイクル数を減らしてください。PCRの線形増幅領域内でPCR産物を解析することが非常に重要です。そうしなければ、増幅前のDNA量の差が正確に測定できません。 |
| 7. 解析したいターゲットの抗体の免疫沈降産物で、PCR反応による増幅が起こらない。 | PCR反応に加えたDNA量が十分でない。 免疫沈降に使用した抗体量が十分でない。 免疫沈降ではワークしない抗体である。 | PCR反応により多くのDNAを使用するか、サイクル数を増加させてください。 通常は抗体1-5 μgを免疫沈降に加えます。しかし、実際に必要な量は抗体によって大きく異なります。 免疫沈降反応液に加える抗体の量を増やしてください。別の抗体を検討してください。 |
更新:2011年12月
改訂日:2022年4月
Protocol Id: 82
ヒト
Polyclonal antibodies are produced by immunizing animals with a synthetic peptide corresponding to amino acid sequence at the N-terminus of human nanog. Antibodies are purified by Protein A and peptide affinity chromatography.
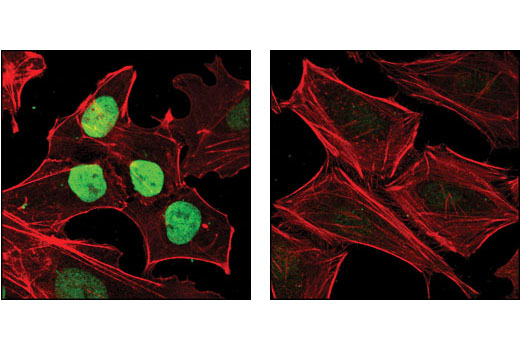
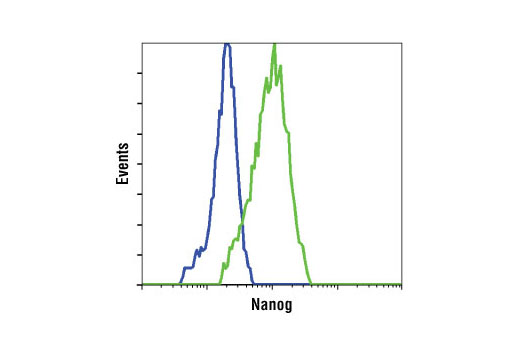
Nanog is a homeodomain-containing transcription factor that is essential for the maintenance of pluripotency and self renewal in embryonic stem cells (1). Nanog expression is controlled by a network of factors including Sox2 and the key pluripotency regulator Oct-4 (1). Recent advances in somatic cell reprogramming have utilized viral expression of combinations of transcription factors including nanog, Oct-4, Sox2, KLF4, c-Myc, and LIN28 (2,3).
Explore pathways related to this product.
STRING - Known and Predicted Protein-Protein Interactions.
Except as otherwise expressly agreed in a writing signed by a legally authorized representative of CST, the following terms apply to Products provided by CST, its affiliates or its distributors. Any Customer's terms and conditions that are in addition to, or different from, those contained herein, unless separately accepted in writing by a legally authorized representative of CST, are rejected and are of no force or effect.
Products are labeled with For Research Use Only or a similar labeling statement and have not been approved, cleared, or licensed by the FDA or other regulatory foreign or domestic entity, for any purpose. Customer shall not use any Product for any diagnostic or therapeutic purpose, or otherwise in any manner that conflicts with its labeling statement. Products sold or licensed by CST are provided for Customer as the end-user and solely for research and development uses. Any use of Product for diagnostic, prophylactic or therapeutic purposes, or any purchase of Product for resale (alone or as a component) or other commercial purpose, requires a separate license from CST. Customer shall (a) not sell, license, loan, donate or otherwise transfer or make available any Product to any third party, whether alone or in combination with other materials, or use the Products to manufacture any commercial products, (b) not copy, modify, reverse engineer, decompile, disassemble or otherwise attempt to discover the underlying structure or technology of the Products, or use the Products for the purpose of developing any products or services that would compete with CST products or services, (c) not alter or remove from the Products any trademarks, trade names, logos, patent or copyright notices or markings, (d) use the Products solely in accordance with CST Product Terms of Sale and any applicable documentation, and (e) comply with any license, terms of service or similar agreement with respect to any third party products or services used by Customer in connection with the Products.
View in English?